AAIC 2018
USE OF COMMON PAIN RELIEVING DRUGS CORRELATES WITH ALTERED PROGRESSION OF ALZHEIMER’S DISEASE AND MILD COGNITIVE IMPAIRMENT
Jack Rivers-Auty , Alison E. Mather , Ruth Peters , Catherine B. Lawrence , David Brough,
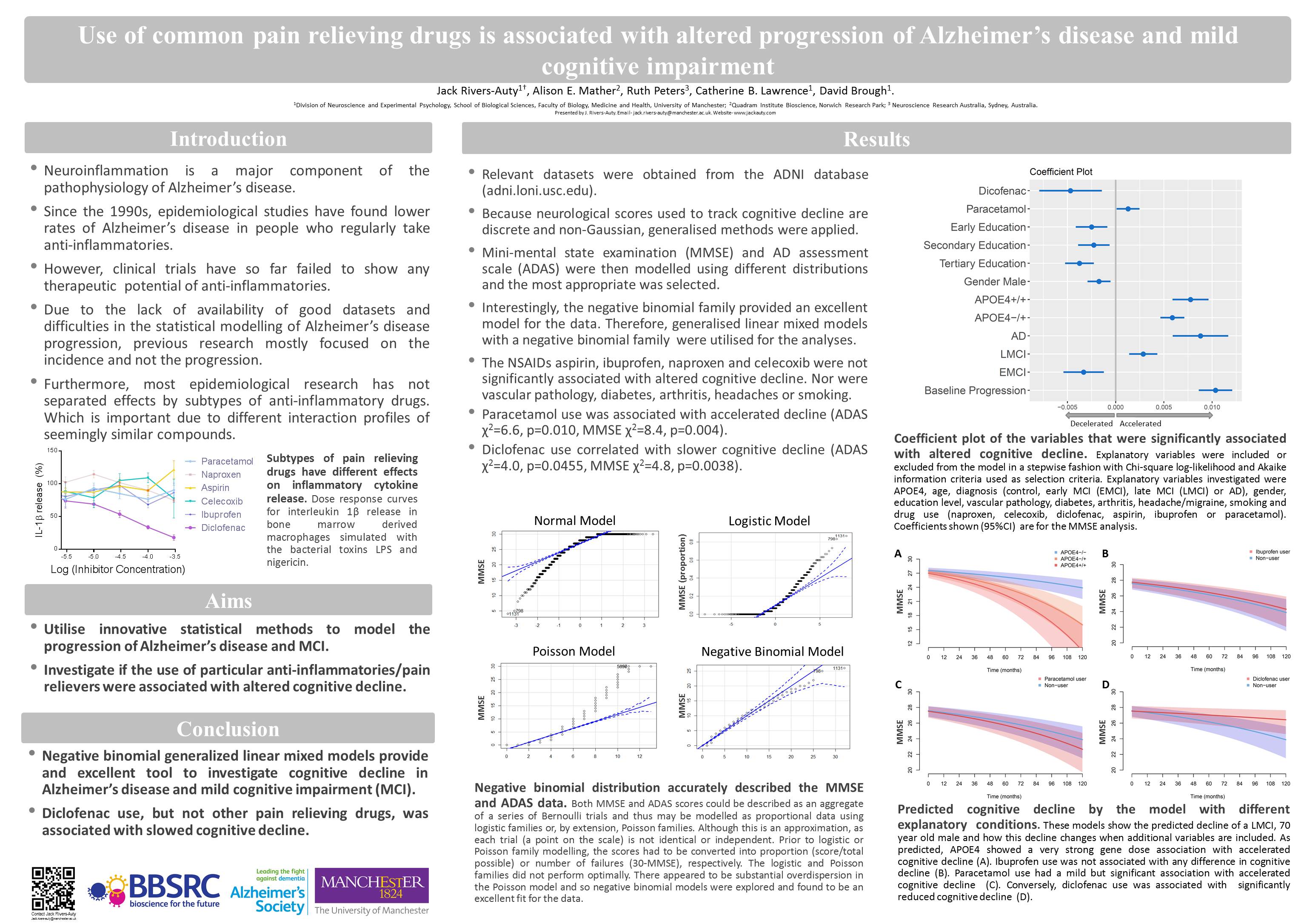
Background: Our understanding of the pathophysiological mechanisms of Alzheimer’s disease (AD) remains relatively unclear; however, the role of neuroinflammation as a key etiological feature is now widely accepted due to the consensus of epidemiological, neuroimaging, preclinical and genetic evidence. Consequently, non-steroidal anti-inflammatories (NSAIDs) have been investigated in epidemiological and clinical studies as potential disease modifying agents. Previous epidemiological studies focused on incidence of AD and did not thoroughly parse the effects at the individual drug level. The therapeutic potential of modifying incidence has a number of limitations, and we now know that each NSAID subtype has a unique profile of physiological impacts corresponding to different therapeutic profiles for AD. Therefore, we utilized the AD Neuroimaging Initiative (ADNI) dataset to investigate how the use of common NSAIDs and paracetamol alter cognitive decline in subjects with mild cognitive impairment (MCI) or AD.
Methods: Negative binomial generalized linear mixed modelling was utilized to model the cognitive decline of 1619 individuals from the ADNI dataset. Both the mini-mental state examination (MMSE) and AD assessment scale (ADAS) were investigated. Explanatory variables were included or excluded from the model in a stepwise fashion with Chi-square log-likelihood and Akaike information criteria used as selection criteria. Explanatory variables investigated were APOE4, age, diagnosis (control, MCI or AD), gender, education level, vascular pathology, diabetes and drug use (naproxen, celecoxib, diclofenac, aspirin, ibuprofen or paracetamol).
Results: The NSAIDs, aspirin, ibuprofen, naproxen and celecoxib did not significantly alter cognitive decline. However, diclofenac use correlated with slower cognitive decline (ADAS χ2= 4.0, p=0.0455, MMSE χ2= 4.8, p=0.029). Paracetamol use correlated with accelerated decline (ADAS χ2= ¼6.6, p=0.010, MMSE χ2= 8.4, p=0.004). The APOE4 allele correlated with accelerated cognitive deterioration (ADAS χ2= 316.0, p<0.0001, MMSE χ2= 191.0, p<0.0001).
Conclusions: This study thoroughly investigated the effects of common NSAIDs and paracetamol on cognitive decline in MCI and AD subjects. Most common NSAIDs did not alter cognitive decline. However, diclofenac use correlated with slowed cognitive deterioration, providing exciting evidence for a potential disease modifying therapeutic. Conversely, paracetamol use correlated with accelerated decline; which, if confirmed to be causative, would have massive ramifications for the recommended use of this prolific drug.
ARUK 2017
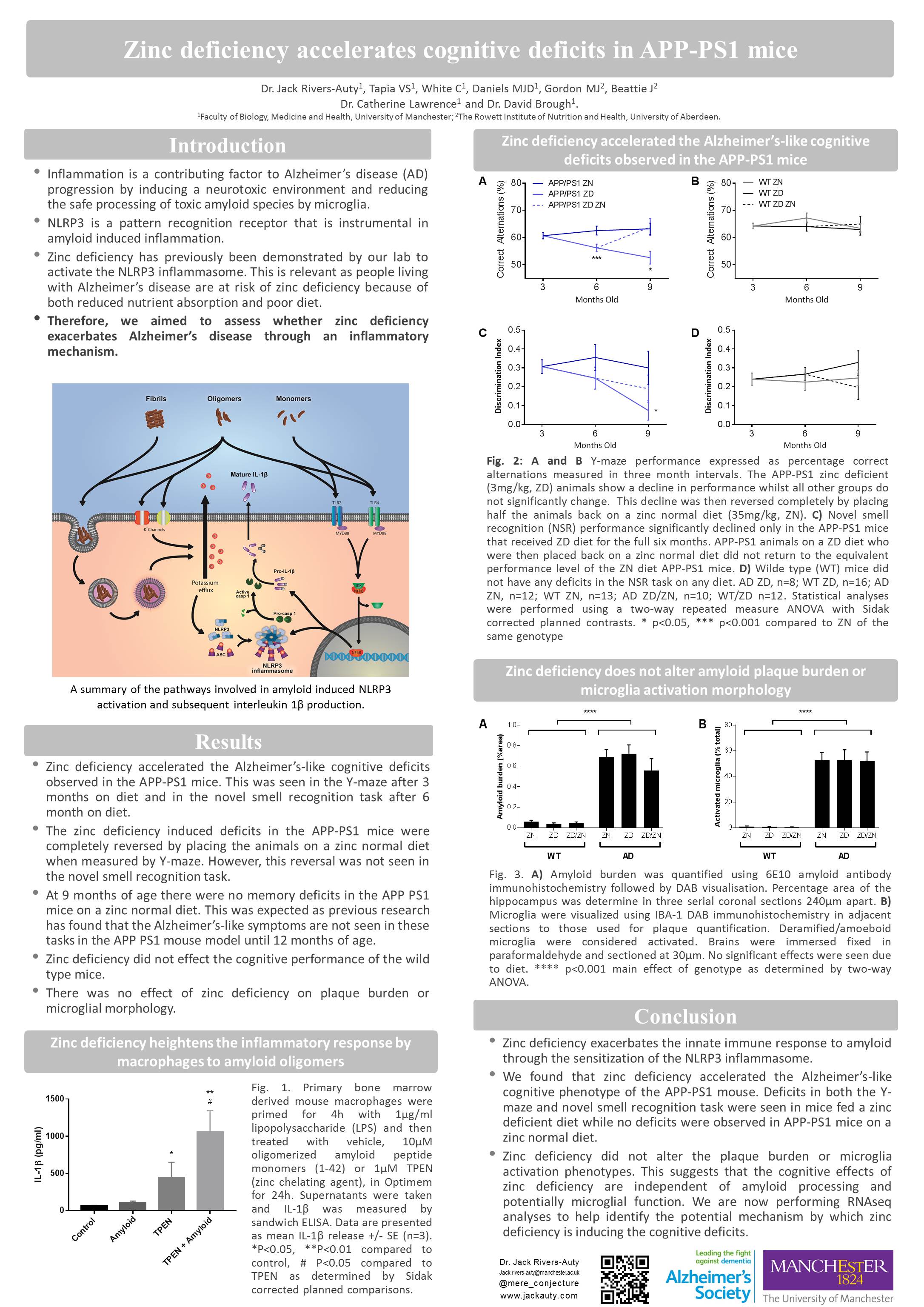
Zinc deficiency accelerates Alzheimer’s phenotype in the APP/PS1 mouse model.
Rivers-Auty J, White C, Beattie J, Brough D, and Lawrence C.
The risk of developing Alzheimer’s disease (AD) increases dramatically as we age. Similarly, the common malnutrition of zinc deficiency is also more prevalent in the elderly more due to changes in diet and zinc absorption as we age. We have shown that zinc deficiency induces the secretion of the inflammatory cytokine interleukin-1β (IL-1β) in macrophages and microglia. This is due to the activation of the IL-1β regulatory protein complex the NLRP3 inflammasome. Neuroinflammation has long been suspected as an exacerbating factor in AD pathology and recently the NLRP3 pattern recognition receptor (PRR) has been found to be the critical in regulated neuroinflammation in mouse models of AD. It is thought that AD associated toxic amyloid oligomers cause disruption in microglia physiology which initiates the formation of the multiprotein NLRP3 inflammasome complex causing the secretion of the IL-1 β. Therefore, this study aimed to assess whether a zinc deficient diet would accelerate the AD behavioural phenotype seen in the APPswe/PS1 mouse model of AD by inducing inflammation through NLRP3 activation. To test this hypothesis, mice were placed on a zinc deficient (3mg/kg) or a zinc normal (35mg/kg) diet for six months. Cognitive/memory performance was evaluated with the Morris water maze, Y-maze and novel smell tasks at baseline, three and six months on diet. To assess if the effect of zinc deficiency was reversible, at the 3 month time-point half the zinc deficient mice were placed back on a zinc normal diet. Our results suggest that zinc deficiency caused memory deficits in the APPswe/PS1 mice at 3 and 6 months on diet while at the same age the wild type mice on a zinc deficient diet and the APPswe/PS1 mice on normal diet showed no deficits. Furthermore, placing the zinc deficient mice on a zinc normal diet partially reverse the memory deficits. Additionally, it was found that innate immune cells from the zinc deficient mice had a hyper-inflammatory phenotype. Therefore, we have shown that zinc deficiency accelerates the AD phenotype in the APPswe/PS1 mice potentially through an inflammatory mechanism. This research aims to further investigate the role of zinc deficiency and NLRP3 in AD using NLRP3-/- mice and novel inhibitors of NLRP3 activation.
AWCBR 2011
Cannabinoid receptor type II as a target for neuroprotection following hypoxia ischemia. Or is it an active vehicle in the driving seat?
J. Rivers and J. C. Ashton
Active marijuana extracts and synthetic analogues, referred to as cannabinoids, elicit their effects through two described cannabinoid receptors. Cannabinoid receptor type 1 (CBI) found ubiquitously throughout the central nervous system (CNS), and cannabinoid receptor type 2 (CBII) found primarily in systemic immune cells. Selective agonists of the CBII receptor produce an immunosuppressant effect without any of the psycho-active effects seen during CBI receptor activation. Some research suggests that administration of CBII agonists before neurological injury may offer some neuroprotection through an anti-inflammatory mechanism. Our research aimed to investigate whether a clinically significant paradigm, of post injury drug administration, would offer neuroprotection in a rat model of early childhood ischemic insult. The model Variable hypoxia ischemia (VHI) was established in our lab and involves a unilateral ligation of the left carotid artery, followed by a period of hypoxia (8%oxygen 72%nitrogen) until a clonic tonic seizure was induced. Drug administration began 30 minutes after VHI and infarction volumes were assessed 15 days after VHI. No neuroprotection was seen when a selective CBII partial agonist (GW 405833) or a selective CBII full agonist (HU 910) was administered using either a single dose of 3mg/kg or with 6 daily doses of 10mg/kg. Unexpectedly one of vehicles used in this study that contained cyclodextrin 25% (w/v) was found to be neuroprotective with a 30.7% reduction in infarction volume when compared to a no vehicle control. This finding is consistent with research that suggests that cyclodextrin reduces excitotoxicity by lowering cholesterol levels at the synaptic cleft, thereby altering glutamate receptivity.

WCBR 2010
Effect of the Cannabinoid CBII Receptor Partial Agonist Gw405833 on Neurological Damage Caused by Hypoxia Ischemia in Rats
Jack Rivers, John Ashton
Two G-protein coupled cannabinoid receptors have been described in mammals. Cannabinoid receptor type 1 (CBI) is ubiquitously expressed in the central nervous system (CNS), whereas cannabinoid receptor type 2 (CBII) is expressed chiefly in systemic immune cells. Cannabinoids that activate both receptors have been long known to have therapeutic effects that include suppression of inflammation and pain, and regulation of nausea, emesis and appetite. More recently, cannabinoids have been shown to be neuroprotective in various animal models. However, the use of cannabinoids as therapeutic drugs has been limited by their psychoactive side-effects, caused by activation of CBI. Following the recent discovery of CBII receptor expression in the CNS, together with evidence to suggest that activation of the CBII receptor causes few or no psychoactive effects, we have investigated whether specific CBII activation is neuroprotective. We therefore tested whether a CBII selective partial agonist GW405833 is neuroprotective in a rat model of hypoxia-ischemia (HI). 26 day old rats (n = 7-8) were administered with 3mg/kg GW405833 or vehicle (i.p) either before or immediately following HI. Neurological damage was assessed at 3 days and 15 days following treatment. Rats administered GW405833 had a reduction in the loss of hemisphere at 15 days post HI (vehicle 8.4% +–0.89% loss of volume, 3 mg/kg GW405833 4.1%. +– 0.64% loss of volume, P=0.001). Our results demonstrate that CBII activation is neuroprotective in the chronic phase of injury following HI and that CBII is a potential target for the treatment of cerebral ischemia.
http://www.conferences.uiuc.edu/WCBR/WCBR2010ProgramForWeb.pdf
