There is growing evidence for the role inflammation in Alzheimer’s disease progression. Genome wide association studies (GWAS) have found that variants of genes involved in regulating innate immune function confer a greater risk in developing Alzheimer’s disease(1 & 2). Examples include loss/reduction of function mutations in the anti-inflammatory/phagocytosis TREM-2 gene (3); variants of promotor regions of inflammation modulating cytokines interleukin-10 (IL-10) and TNFα (4); and variants of the anti-inflammatory/phagocytosis receptor CD33 (5). The number and range of risk genes that are related to Alzheimer’s disease demonstrate the integral role inflammation plays in the pathogenesis of Alzheimer’s disease. There are multiple pathways by which inflammation is induced and regulated in Alzheimer’s disease, therefore, pharmacologically targeting multiple key inflammatory pathways should confer the greatest therapeutic effect(6).
Epidemiological evidence suggests that chronic non-steroidal anti-inflammatories (NSAIDs) use corresponds to significantly lower rates of Alzheimer’s disease (7,8). This evidence resulted in several small, short term, clinical trials of NSAIDs which had largely negatively results showing no differences in cognitive measures compared to placebo control (9). The failure of these trials has previously been attributed to the designs of the clinical trials or hidden biases in the epidemiological studies. However, our group has shown that the NSAIDs used in the clinical trials were perhaps too specific to the cyclooxygenase (COX)/eicosanoids inflammatory pathway, leaving other potentially compensatory inflammatory pathways unaffected (10). One example of a COX-independent inflammatory pathway involves the NLRP3 inflammasome.
Recent studies using knock-out (KO) mice have identified the pattern recognition receptor (PRR) NLRP3 as a crucial mediator of neuroinflammation in animal models of Alzheimer’s disease (11,12) (Figure 1). The NLRP3 protein recognizes a number of sterile stimuli, including amyloid oligomers (13), and once activated oligomerizes with the ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) protein to form the NLRP3 inflammasome, which in turn recruits procaspase-1 causing its activation (14,15) (Figure 1). Caspase-1 then cleaves prointerleukin-1β (pro-IL-1β) into interleukin-1β (IL-1β), which is then released by the cell where it can initiate an inflammatory response through the IL-1 receptor 1 (14,15) (Figure 1). Non-fenamate NSAIDs do not inhibit this inflammatory pathway which is thought to be active in Alzheimer’s disease. Therefore, the failure of NSAIDs in clinical trials may be partially attributed to unimpeded neuroinflammation mediated through NLRP3 activity. This hypothesis was researched by myself, Michael Daniels, Dr. David Brough and others and the result were recently published in Nature Communications (11).
Dr. David Brough enlisted a fantastic Ph.D. student Michael Daniels to investigate if non-classical NSAIDs had any inhibitory activity on the NLRP3 inflammasome and they found for the first time that the fenamate subclass of NSAIDs are polyvalent drugs which inhibit NLRP3 activation in addition to COX activity (11). This suggested to us that fenamate NSAIDs may provide a drastic therapeutic effect in diseases in which sterile inflammation is a contributing factor, such as Alzheimer’s disease.
We therefore tested the fenamate NSAID mefenamic acid in an amyloid injection rat model related to Alzheimer’s disease and a genetic mouse model of Alzheimer’s disease. From this we found that mefenamic acid prevented memory loss in the rat model and reversed memory loss in the mouse model of AD. As well as dramatically reducing inflammation in the brains of the Alzheimer’s diseased mice.
This is preclinical research and further work is needed including corroborative preclinical research and clinical studies before we can conclusively say what effects fenamate NSAIDs have on Alzheimer’s Disease progression. However, it is a very exciting result which we will continue to research. We are particularly excited about the future of fenamates because these drugs have been used in the clinic for decades for pain relief, which means that have an established safety profile, reducing the cost and time it takes to validate these drugs’ potential as an Alzheiemer’s treatments in the clinic.
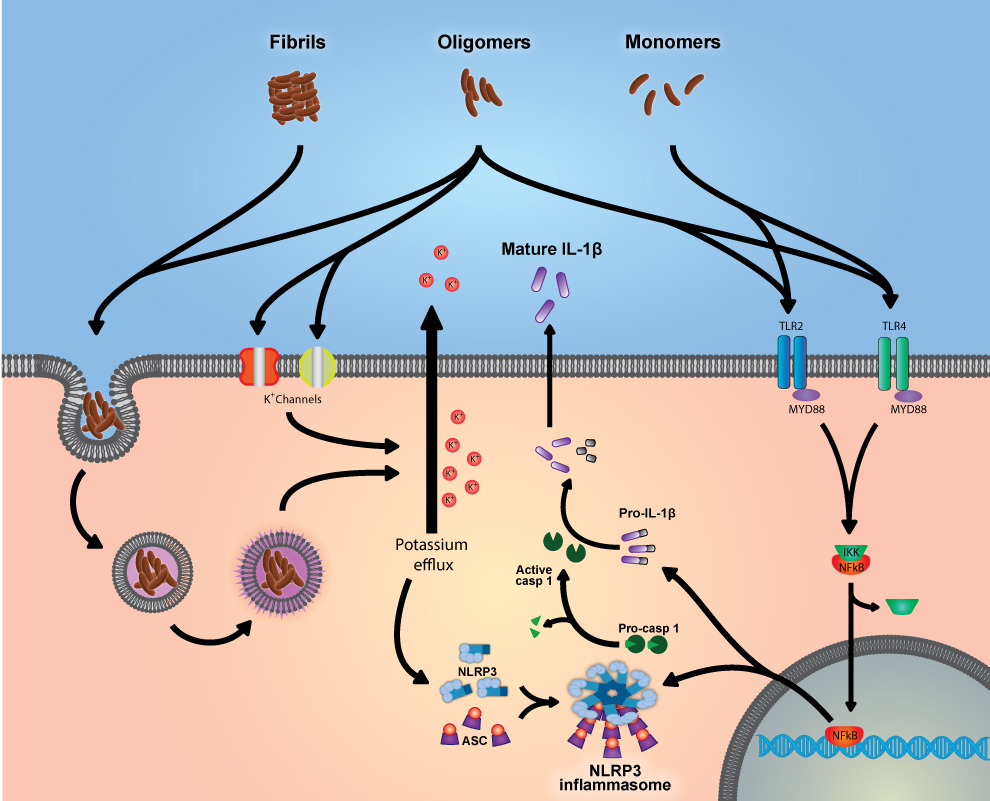 Figure 1: Amyloid monomers, oligomers and fibrils interact with a number of cellular pathways to cause the expression of the components of the NLRP3 inflammasome and proIL-1β, as well as activating the nucleation of the NLRP3 inflammasome leading to the production of mature IL-1β.
Figure 1: Amyloid monomers, oligomers and fibrils interact with a number of cellular pathways to cause the expression of the components of the NLRP3 inflammasome and proIL-1β, as well as activating the nucleation of the NLRP3 inflammasome leading to the production of mature IL-1β.
- Heppner, F.L., Ransohoff, R.M. & Becher, B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358-72 (2015).
- Ramanan, V.K. & Saykin, A.J. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. American Journal of Neurodegenerative Disease 2, 145-175 (2013).
- Guerreiro , R. et al. TREM2 Variants in Alzheimer’s Disease. New England Journal of Medicine 368, 117-127 (2013).
- Ramos, E.M. et al. Tumor necrosis factor alpha and interleukin 10 promoter region polymorphisms and risk of late-onset Alzheimer disease. Arch Neurol 63, 1165-9 (2006).
- Bradshaw, E.M. et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci 16, 848-850 (2013).
- Heneka, M.T., Golenbock, D.T. & Latz, E. Innate immunity in Alzheimer’s disease. Nat Immunol 16, 229-236 (2015).
- Breitner, J.C. et al. Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging 16, 523-30 (1995).
- McGeer, P.L., Schulzer, M. & McGeer, E.G. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 47, 425-432 (1996).
- Miguel-Alvarez, M. et al. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging 32, 139-47 (2015).
- Minghetti, L. Cyclooxygenase-2 (COX-2) in Inflammatory and Degenerative Brain Diseases. Journal of Neuropathology & Experimental Neurology 63, 901-910 (2004).
- Daniels, M.J.D†. and Rivers-Auty†, J. et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in mice. Nature Communications ( †equal contributors).
http://www.nature.com/ncomms/2016/160811/ncomms12504/full/ncomms12504.html - Heneka, M.T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674-8 (2013).
- Parajuli, B. et al. Oligomeric amyloid [beta] induces IL-1[beta] processing via production of ROS: implication in Alzheimer/’s disease. Cell Death Dis 4, e975 (2013).
- Baroja-Mazo, A. et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15, 738-748 (2014).
- Franklin, B.S. et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15, 727-737 (2014).